











Forschende der Universität Bonn haben ein KI-Verfahren so trainiert, dass sich damit potenzielle Wirkstoffe mit besonderen Eigenschaften vorhersagen lassen. Dazu nutzten sie ein chemisches Sprachmodell - eine Art ChatGPT für Moleküle. Nach einer Trainingsphase konnte die KI die chemischen Strukturformeln von Verbindungen erzeugen, die sich möglicherweise als besonders wirksame Medikamente eignen. Die Studie ist nun in Cell Reports Physical Science erschienen.
Wer die Oma zu ihrem 90. Geburtstag mit einem Gedicht erfreuen möchte, muss heutzutage kein Poet sein: Ein kurzer Prompt bei ChatGPT genügt, und binnen weniger Sekunden spuckt die KI eine lange Liste von Wörtern aus, die sich auf den Namen der Jubilarin reimen. Auf Wunsch erzeugt sie dazu sogar ein Sonett.
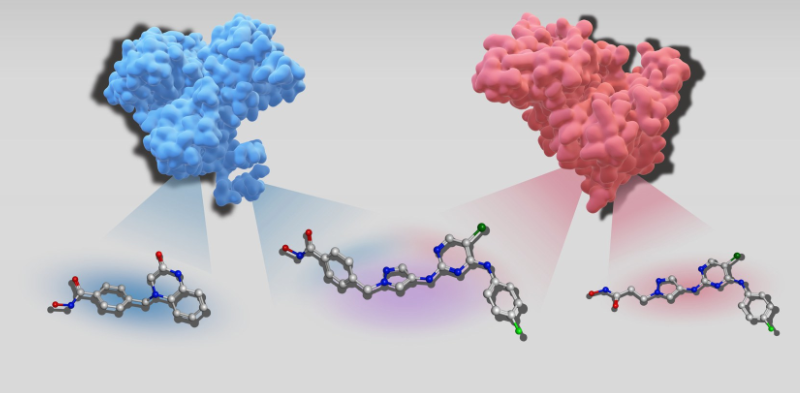
Forschende der Universität Bonn haben in ihrer Studie ein ähnliches Modell implementiert - ein sogenanntes chemisches Sprachmodell. Damit lassen sich allerdings keine Reime produzieren. Stattdessen gibt die KI die Strukturformeln chemischer Verbindungen aus, die möglicherweise eine besonders begehrenswerte Eigenschaft aufweisen: Sie sind dazu in der Lage, an zwei unterschiedliche Zielproteine zu binden. Im Organismus können sie so zum Beispiel gleichzeitig zwei Enzyme hemmen.
Gesucht: Wirkstoffe mit dem „Doppel-Wumms“
„In der Pharmaforschung sind derartige Wirkstoffe aufgrund ihrer Polypharmakologie sehr begehrt“, erläutert Prof. Dr. Jürgen Bajorath. Der Chemieinformatiker leitet am Lamarr-Institut für maschinelles Lernen und künstliche Intelligenz den Bereich KI in den Lebenswissenschaften sowie am b-it (Bonn-Aachen International Center for Information Technology) der Uni Bonn das Life Science Informatics-Programm. „Da sie mehrere intrazelluläre Prozesse und Signaltransduktionswege zugleich beeinflussen, sind sie oft besonders wirksam - etwa im Kampf gegen Krebs.“ Im Prinzip lässt sich dieser Effekt zwar auch durch die Kombination verschiedener Präparate erreichen. Dabei riskiert man aber Wechselwirkungen zwischen den einzelnen Medikamenten. Außerdem werden verschiedene Verbindungen meist unterschiedlich schnell abgebaut, was ihre gemeinsame Verabreichung erschwert.
Ein Molekül zu finden, das die Wirkung eines einzelnen Zielproteins spezifisch beeinflusst, ist schon nicht einfach. Umso komplizierter ist es, Verbindungen zu designen, die einen „Doppel-Wumms“ entfalten. Chemische Sprachmodelle können dabei zukünftig möglicherweise helfen. ChatGPT wird mit Milliarden Seiten von geschriebenen Texten trainiert und lernt daraus, selbst Sätze zu formulieren. Chemische Sprachmodelle funktionieren ähnlich, haben aber nur vergleichsweise kleine Datenmengen zur Verfügung. Aber auch sie werden im Prinzip mit Texten gefüttert, zum Beispiel den sogenannten SMILES-Strings, die organische Moleküle und deren Struktur als eine Sequenz von Buchstaben und Symbolen darstellen. „Wir haben unser chemisches Sprachmodell nun mit Paaren von Strings trainiert“, sagt Sanjana Srinivasan aus Bajoraths Arbeitsgruppe. „Einer davon beschrieb jeweils ein Molekül, von dem wir wissen, dass es nur gegen ein Zielprotein wirkt. Der zweite stand dagegen für eine Verbindung, die neben diesem Protein zusätzlich noch ein zweites Zielprotein beeinflusst.“
KI lernt chemische Zusammenhänge
Das Modell wurde mit mehr als 70.000 dieser Paare gefüttert. Auf diese Weise eignete es sich ein implizites Wissen darüber an, worin sich die normalen Wirkstoffe von denen mit dem Doppel-Wumms unterschieden. „Wenn wir es danach mit einer Verbindung gegen ein Zielprotein fütterten, schlug es auf dieser Basis Moleküle vor, die nicht nur gegen dieses Protein, sondern auch noch ein weiteres wirken sollten“, erklärt Bajorath.
Die Trainings-Verbindungen mit dem Doppel-Wumms richten sich oft gegen Proteine, die sich ähneln und die dementsprechend im Körper eine ähnliche Funktion übernehmen. In der Pharmaforschung fahndet man aber auch nach Wirkstoffen, die völlig unterschiedliche Klassen von Enzymen oder Rezeptoren beeinflussen. Um die KI auf diese Aufgabe vorzubereiten, erfolgte nach der generellen Lernphase noch ein Feintuning. Darin brachten die Forschenden dem Algorithmus mit Hilfe von ein paar Dutzend speziellen Trainings-Paaren bei, gegen welche unterschiedlichen Proteinklassen sich die vorgeschlagenen Verbindungen richten sollten. Das ist in etwa so, als würde man ChatGPT instruieren, diesmal kein Sonett zu erzeugen, sondern einen Limerick.
Tatsächlich spuckte das Modell nach dem Feintuning Moleküle aus, bei denen bereits nachgewiesen wurde, dass sie gegen die gewünschten Kombinationen von Zielproteinen wirken. „Das zeigt, dass das Verfahren funktioniert“, sagt Bajorath. Die Stärke des Ansatzes ist seiner Meinung nach aber nicht, dass sich damit auf Anhieb neue Verbindungen finden lassen, die die verfügbaren Pharmaka in ihrer Wirkung übertreffen. „Interessanter ist aus meiner Sicht, dass die KI oft chemische Strukturen vorschlägt, an die die meisten Chemiker auf Anhieb gar nicht denken würden“, erklärt er. „Sie generiert gewissermaßen ‚out-of-the-box‘-Ideen und kommt so auf originelle Lösungen, die die Pharma-Forschung zu neuen Ansätzen inspirieren können.“
Publikation
Sanjana Srinivasan and Jürgen Bajorath: Generation of Dual-Target Compounds Using a Transformer Chemical Language Model; Cell Reports Physical Science; DOI: 10.1016/j.xcrp.2024.102255; https://doi.org/10.1016/j.xcrp.2024.102255
Kontakt
Prof. Dr. Jürgen Bajorath
Life Science Informatics
Tel. 0228/73-69100
E-Mail: bajorath@bit.uni-bonn.de