Genomik & Immunregulation
Prof. Dr. med. Joachim L. Schultze


Shiny-Seq: advanced guided transcriptome analysis
A comprehensive analysis of RNA-Seq data uses a wide range of different tools and algorithms, which are normally limited to R users only. While several tools and advanced analysis pipelines are available, some require programming skills and others lack the support for many important features that enable a more comprehensive data analysis. There is thus, a need for a guided and easy to use comprehensive RNA-Seq data platform, which integrates the state of the art analysis workflow.
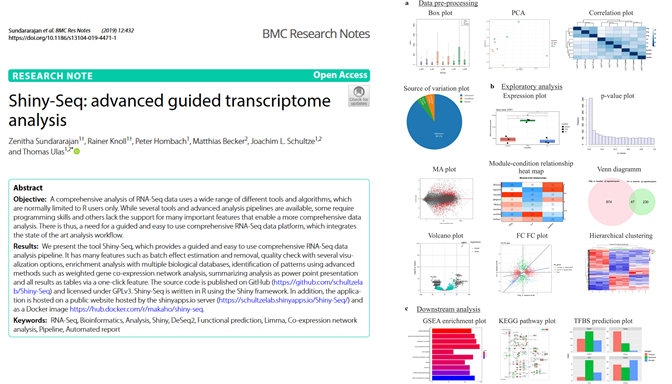
We present the tool Shiny-Seq, which provides a guided and easy to use comprehensive RNA-Seq data analysis pipeline. It has many features such as batch effect estimation and removal, quality check with several visualization options, enrichment analysis with multiple biological databases, identification of patterns using advanced methods such as weighted gene co-expression network analysis, summarizing analysis as power point presentation and all results as tables via a one-click feature. The source code is published on GitHub (https://github.com/schultzelab/Shiny-Seq) and licensed under GPLv3. Shiny-Seq is written in R using the Shiny framework. In addition, the application is hosted on a public website hosted by the shinyapps.io server (https://schultzelab.shinyapps.io/Shiny-Seq/) and as a Docker image https://hub.docker.com/r/makaho/shiny-seq.
For further details on the functions of Shiny-Seq please refer to our latest publication on Shiny-Seq: https://bmcresnotes.biomedcentral.com/track/pdf/10.1186/s13104-019-4471-1.pdf